Clinical Presentation
WN, age 53, presented after 3 weeks of numbness and pain in the right arm, both legs, and perineum. They had no prior medical history. Initially, WN developed insidious onset of numbness in the right arm, mostly involving the entire hand. Within several days, they noted numbness in both legs and the groin with a painful cramping and burning sensation in both legs, which was worse on the left side. WN also reported some mid-back pain and difficulty with balance. They did not report any weakness; urinary, bowel, or sexual dysfunction; fevers; or other systemic symptoms.
On neurologic exam, WN’s mental status and cranial nerve function were intact, but sensation to temperature and pinprick was decreased in the right hand and left leg in a patchy, nondermatomal distribution. WN had full muscular strength on confrontation testing, with 2+ reflexes noted in the upper extremities and 3+ at the patella bilaterally with crossed adduction. They exhibited an extensor plantar response on the left side and a positive Romberg sign. WN was able to ambulate normally but struggled with tandem gait.
WN’s sensory deficits, which were distributed in a nondermatomal fashion in the left leg and right arm, suggested bilateral spinothalamic tract or multiple nerve involvement, whereas perineal numbness implied spinal cord or nerve root localization–possibly involving the conus medullaris or cauda equina. The positive Romberg sign suggested dorsal column dysfunction. WN’s upper motor neuron signs, with hyperreflexia of the lower extremities and an upgoing toe on the left side, implicated the corticospinal tracts (left >right) and disfavored peripheral processes such as Guillain-Barré syndrome. Overall, WN’s presentation pointed to a spinal cord process as the most likely etiology of the constellation of symptoms.
Diagnostic Studies and Initial Treatment
Cervical, thoracic, and lumbar spine MRI with and without contrast revealed multifocal short-segment T2 hyperintense lesions, diffusely central in location and involving both gray and white matter (Figure). None of the lesions were longitudinally extensive, although some showed trace areas of contrast enhancement. Enhancement in the cauda equina nerve roots and focal lobular enhancement of the left S1 nerve root was observed. Brain MRI showed several small T2-hyperintense brainstem lesions, none of which displayed contrast enhancement. These radiologic findings were most consistent with an acute or subacute inflammatory process.
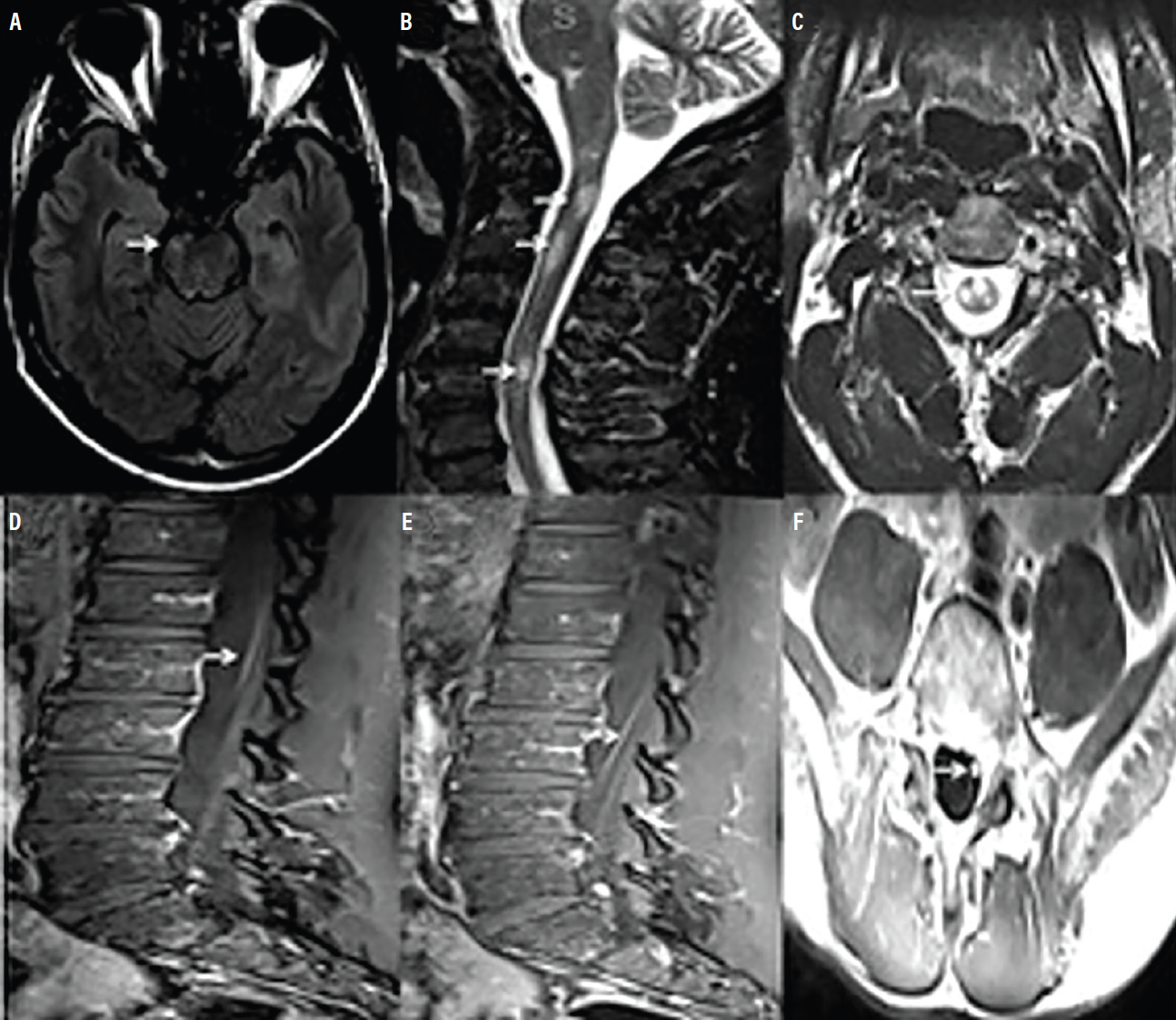
Figure 1 (A-F): Representative brain and spine MRI findings are shown. A small T2 hyperintense brainstem lesion is viewed axially (A). Cervical spine sagittal (B) and axial (C) views show multiple short-segment T2 hyperintense lesions. Lumbar spine sagittal (D, E) views of T1 demonstrate post-contrast enhancement of cauda equina nerve roots. Lumbar spine axial (F) view of T1 post-contrast imaging shows focal lobular enhancement of the left S1 nerve root.
Empiric treatment with pulse-dose intravenous (IV) steroids was initiated, and further extensive evaluation continued for potential inflammatory, infectious, and neoplastic causes of their myeloradiculitis.
Serology was remarkable for mildly elevated angiotensin-converting enzyme (ACE; 73 U/L). Results were all normal for eosinophil sedimentation rate (ESR), C-reactive protein (CRP), thyroid stimulating hormone (TSH), serum protein electrophoresis (SPEP), vitamin B12, vitamin E, copper, and zinc. Additionally, tests were negative for HIV, rapid plasma reagin (RPR), Lyme disease, antinuclear antibodies (ANA), extractable nuclear antigens (ENA), rheumatoid factor (RF), human T-lymphocyte-1 (HTLV-1) antibody, and ganglioside antibodies. Serum antiaquaporin 4 antibody (antiAQP4) and myelin oligodendrocyte glycoprotein antibody (antiMOG) tests were ordered, but the results were not available until after WN was discharged from the hospital.
Cerebrospinal fluid (CSF) analysis showed elevated protein (53 mg/dL), normal cell counts (white blood cells [WBC] 4 / mL), and a normal glucose level. Venereal disease research laboratory (VDRL) and ACE tests of CSF were negative. There were no oligoclonal bands. CSF cytology and flow cytometry were unremarkable (other than a slightly elevated CD4:CD8 cell ratio). Meningitis and encephalitis polymerase chain reaction (PCR) panels and cultures of CSF were negative. Chest CT showed no pathologic findings.
Differential Diagnosis
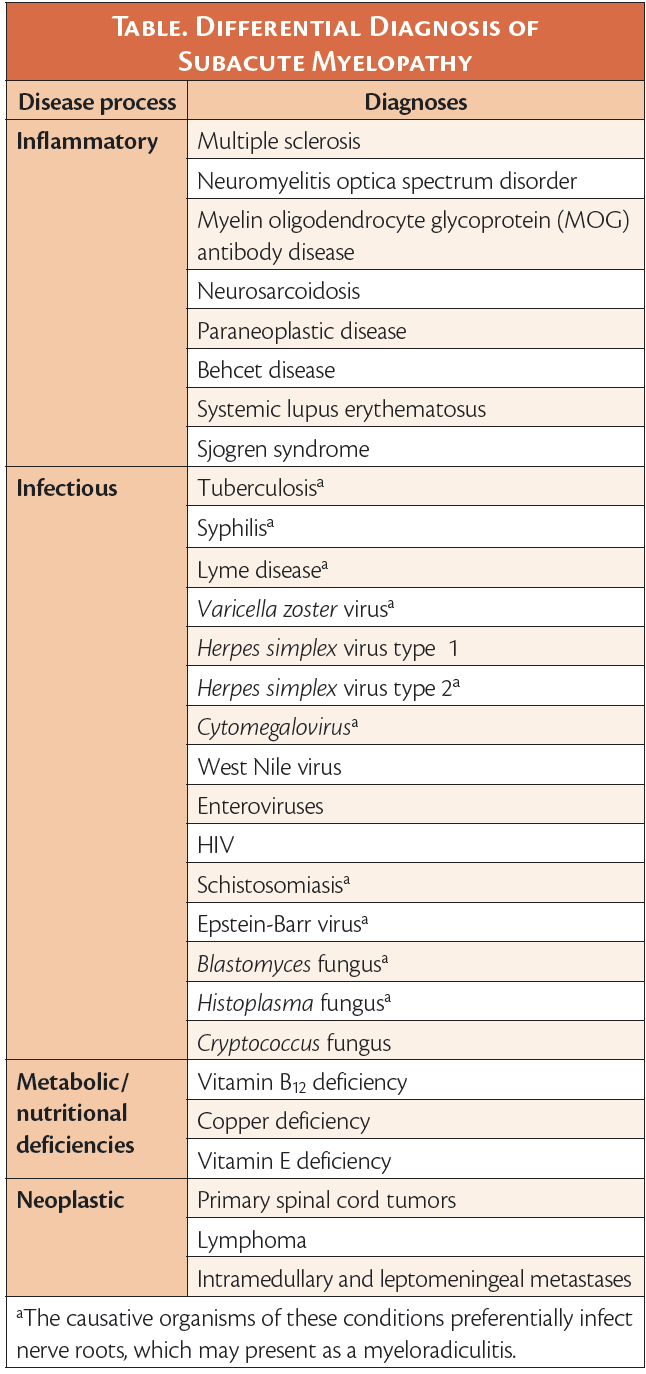
The differential diagnosis of subacute myelopathy broadly includes inflammatory, infectious, and neoplastic processes and metabolic diseases (Table).1-3 A subacute multifocal process involving the spinal cord, nerve roots, and to a lesser extent, the brainstem, without evidence of infection, suggested an inflammatory etiology of myeloradiculitis. Neurosarcoidosis can result in elevated serum ACE, spinal nerve root enhancement, and myelitis, although it usually manifests as longitudinally extensive transverse myelitis (LETM). Also, in neurosarcoidosis, contrast enhancement most frequently occurs in a subpial and/or meningeal pattern.4 WN’s chest CT was negative for pulmonary sarcoidosis. Neuromyelitis optica spectrum disorder (NMOSD) was considered but is also typically associated with isolated LETM and rarely involves spinal nerve roots.5 Moreover, antiAQP4 testing was negative after WN was discharged, ruling out NMOSD. MOG antibody disease (MOGAD) was also considered because of WN’s multifocal demyelinating lesions, especially because lower thoracic cord involvement was observed.
Management and Follow-up
WN had some clinical improvement after 3 days of IV steroid treatment and was discharged with a prescription for prolonged oral steroid taper over 12 weeks. A positive result for serum antiMOG (1:100 by cell-based assay, Mayo Laboratory Clinic, Rochester, MN) was returned after discharge, suggesting a diagnosis of MOGAD. At follow-up 2 weeks later, WN reported further improvement of their numbness and balance problems. A repeat MRI done 8 weeks after hospitalization showed stable areas of spinal nerve root enhancement and a decrease in size of some of the cord lesions. WN elected monthly IV immunoglobulin (IVIG) maintenance therapy and was clinically stable for over 12 months at the time of their last follow-up.
Discussion
Background
MOG is located on the outer surface of the myelin sheath. MOGAD is an inflammatory demyelinating disease that is distinct from multiple sclerosis and antiAQP4+ NMOSD.6 At present, there are no formal diagnostic criteria for MOGAD, which can have a monophasic course (particularly in children) or a relapsing course. Approximately 40% of adults who present with MOGAD have another attack within 5 years. Elevated antiMOG titers are present, particularly in the acute phase of the clinical attack, and may decrease over months following the initial presentation of symptoms.7

Diagnosis
MOGAD has been associated with diverse clinical presentations, including recurrent and bilateral optic neuritis, acute disseminated encephalomyelitis (ADEM) (in children), focal cortical or brainstem encephalitis, and transverse myelitis (TM) with predilection for the conus medullaris. There are higher reported incidences of neurogenic bowel and bladder symptoms and erectile dysfunction associated with MOGAD, likely due to the myelitis more frequently involving the conus medullaris.

Associated spinal cord lesions are often multifocal because MOGAD tends to simultaneously affect multiple areas of the central nervous system (CNS),6 as was the case for WN. Although LETM is more common, short segment transverse myelitis is seen in up to 40% of cases.8 Radiologic signs that can help distinguish MOGAD-associated myelitis from other inflammatory myelitides include minimal or lack of contrast enhancement and longitudinally extensive T2 signal abnormality confined to gray matter, resulting in a T2 hyperintense line on the sagittal view and an “H sign” on the axial view.9

Spinal nerve root involvement has rarely and only recently been described in MOGAD.10-15 A retrospective cohort study characterized 19 people with MOGAD manifesting as peripheral nervous system (PNS) involvement, including 3 individuals with myeloradiculitis. Like WN, these 3 people had CNS involvement during their disease course and were also responsive to immunotherapy.13

Treatment
TM caused by MOGAD typically responds well to steroids and has a more favorable long-term prognosis than NMOSD-induced myelitis.6,7 With severe attacks or inadequate response to steroids, acute escalation of treatment (plasma exchange or IVIG) is sometimes warranted. Evidence from observational studies suggests that weaning steroids slowly may help mitigate the risk of symptom flares.16,17 At present, there are no reliable ways to predict relapse risk in MOGAD. Recent studies suggest that antiMOG titers in adults do not correlate well with relapse risk.18 Some individuals with MOGAD may be persistently seropositive for years and still have a monophasic course, whereas others could relapse despite becoming seronegative for antiMOG for a period of time. Some people with MOGAD may require chronic immunotherapy, and several retrospective studies demonstrate good results after treating relapsing MOGAD, including with monthly IVIG monotherapy, among other immunomodulatory agents.16,19-21 Multiple factors should be considered when creating a long-term treatment plan for a person with MOGAD after their first attack, including the severity of the attack and the degree of therapeutic response, risks of cumulative disability, risks of immunotherapy, and age.7

Summary
The case of myeloradiculitis discussed illustrates that MOGAD can manifest clinically and radiographically in both the CNS and PNS. Although rare, MOGAD should particularly be a diagnostic consideration in cases of acute myelitis with both spinal cord and nerve root enhancement. Testing for antiMOG should be carried out under these circumstances because MOGAD can mimic or closely resemble neurosarcoidosis, which was the presumptive diagnosis for WN until a positive antiMOG test was returned. Although MOGAD tends to initially respond well to steroid treatment, long-term management after a first attack is still debated. We continue to broaden our understanding of the clinical phenotypic spectrum of MOGAD with cases like that of WN.
Acknowledgments
The authors thank Ilya Kister, MD for critical review of the manuscript.
1. Lopez Chiriboga S, Flanagan EP. Myelitis and other autoimmune myelopathies. Continuum (Minneap Minn). 2021;27(1):62-92.
2. Pruitt AA. Neoplastic myelopathies. Continuum (Minneap Minn). 2021;27(1):121-142.
3. Toledano M. Infectious myelopathies. Continuum (Minneap Minn). 2021;27(1):93-120.
4. Murphy OC, Salazar-Camelo A, Jimenez JA, et al. Clinical and MRI phenotypes of sarcoidosis-associated myelopathy. Neurol Neuroimmunol Neuroinflamm. 2020;7(4):e722. doi:10.1212/NXI.0000000000000722
5. Takai Y, Misu T, Nakashima I, et al. Two cases of lumbosacral myeloradiculitis with anti-aquaporin-4 antibody. Neurology. 2012;79(17):1826-1828.
6. Parrotta E, Kister I. The expanding clinical spectrum of myelin oligodendrocyte glycoprotein (MOG) antibody associated disease in children and adults. Front Neurol. 2020;11:960. doi:10.3389/fneur.2020.00960
7. Marignier R, Hacohen Y, Cobo-Calvo A, et al. Myelin-oligodendrocyte glycoprotein antibody-associated disease [published correction appears in Lancet Neurol. 2021 Oct;20(10):e6] [published correction appears in Lancet Neurol. 2022 Jan;21(1):e1]. Lancet Neurol. 2021;20(9):762-772. doi:10.1016/S1474-4422(21)00218-0
8. Ciron J, Cobo-Calvo A, Audoin B, et al. Frequency and characteristics of short versus longitudinally extensive myelitis in adults with MOG antibodies: a retrospective multicentric study. Mult Scler. 2020;26(8):936-944.
9. Dubey D, Pittock SJ, Krecke KN, et al. Clinical, radiologic, and prognostic features of myelitis associated with myelin oligodendrocyte glycoprotein autoantibody. JAMA Neurol. 2019;76(3):301-309.
10. Sundaram S, Nair SS, Jaganmohan D, Unnikrishnan G, Nair M. Relapsing lumbosacral myeloradiculitis: an unusual presentation of MOG antibody disease. Mult Scler. 2020;26(4):509-511.
11. Vazquez Do Campo R, Stephens A, Marin Collazo IV, Rubin DI. MOG antibodies in combined central and peripheral demyelination syndromes. Neurol Neuroimmunol Neuroinflamm. 2018;5(6):e503. doi:10.1212/NXI.0000000000000503
12. Shima T, Tsujino A. MOG antibody-related disease with recurrent optic neuritis and sensory polyradiculoneuropathy: a case report. Mult Scler Relat Disord. 2020;46:102597. doi:10.1016/j.msard.2020.102597
13. Rinaldi S, Davies A, Fehmi J, et al. Overlapping central and peripheral nervous system syndromes in MOG antibody-associated disorders. Neurol Neuroimmunol Neuroinflamm. 2020;8(1):e924. doi:10.1212/NXI.0000000000000924
14. Nakamura T, Kaneko K, Watanabe G, et al. Myelin oligodendrocyte glycoprotein-IgG-positive, steroid-responsive combined central and peripheral demyelination with recurrent peripheral neuropathy. Neurol Sci. 2021;42(3):1135-1138.
15. Kang MS, Kim MK, Kim YE, Kim JH, Kim BJ, Lee HL. Two cases of myelin oligodendrocyte glycoprotein antibody-associated disease presenting with cauda equina syndrome without conus myelitis. Mult Scler Relat Disord. 2021;52:103017.
16. Jarius S, Ruprecht K, Kleiter I, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 2: epidemiology, clinical presentation, radiological and laboratory features, treatment responses, and long-term outcome. J Neuroinflammation. 2016;13(1):280.
17. Ramanathan S, Mohammad S, Tantsis E, et al. Clinical course, therapeutic responses and outcomes in relapsing MOG antibody-associated demyelination. J Neurol Neurosurg Psychiatry. 2018;89(2):127-137.
18. Cobo-Calvo A, Ruiz A, Rollot F, et al. Clinical features and risk of relapse in children and adults with myelin oligodendrocyte glycoprotein antibody-associated disease. Ann Neurol. 2021;89(1):30-41.
19. Cobo-Calvo A, Sepúlveda M, Rollot F, et al. Evaluation of treatment response in adults with relapsing MOG-Ab-associated disease. J Neuroinflammation. 2019;16(1):134.
20. Chen JJ, Flanagan EP, Bhatti MT, et al. Steroid-sparing maintenance immunotherapy for MOG-IgG associated disorder. Neurology. 2020;95(2):e111-e120. doi:10.1212/WNL.0000000000009758
21. Whittam DH, Cobo-Calvo A, Lopez-Chiriboga AS, et al. Treatment of MOG-IgG-associated disorder with rituximab: an international study of 121 patients. Mult Scler Relat Disord. 2020;44:102251. doi:10.1016/j.msard.2020.102251
JP and EC report no financial disclosures


