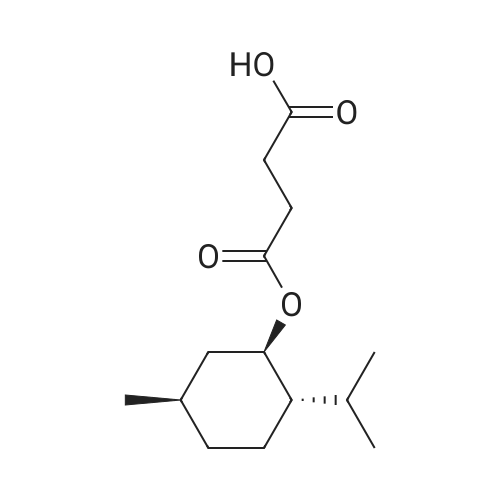
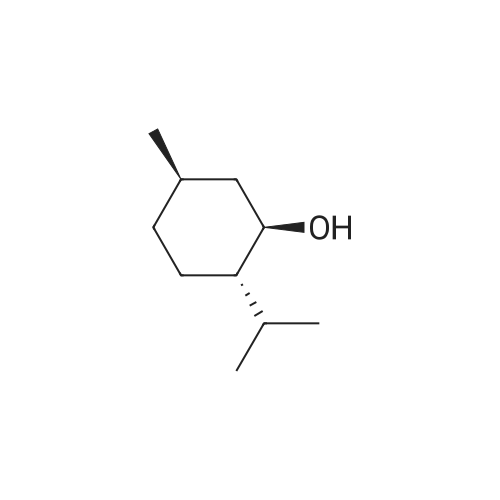
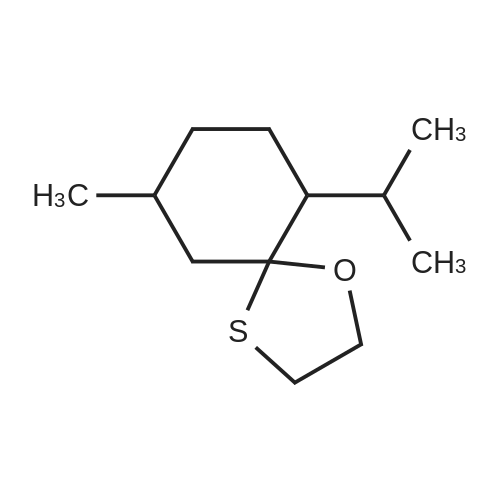
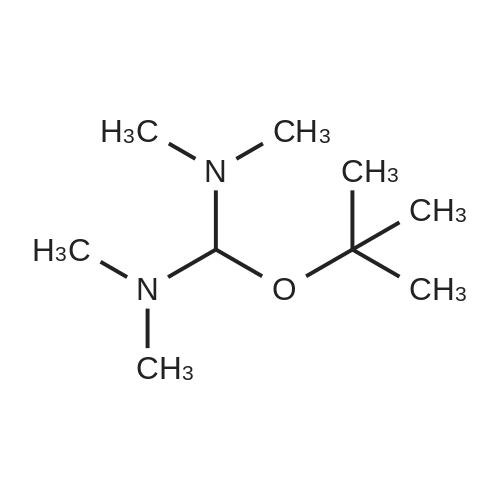
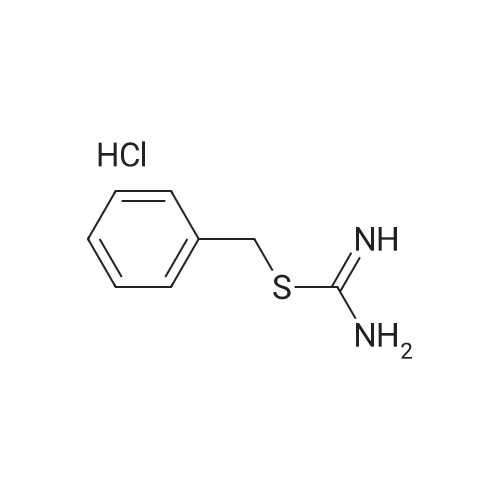
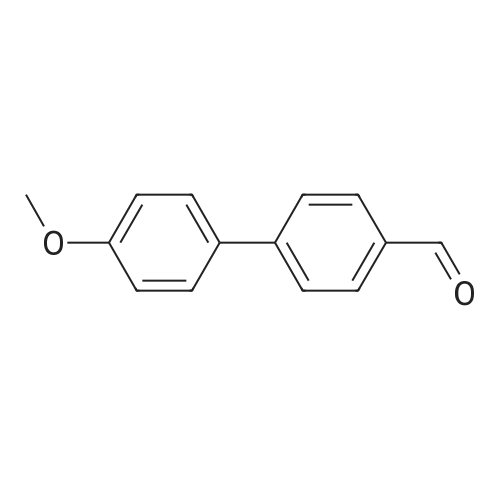
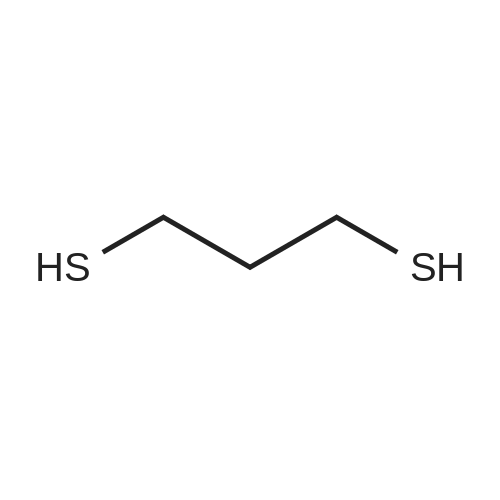
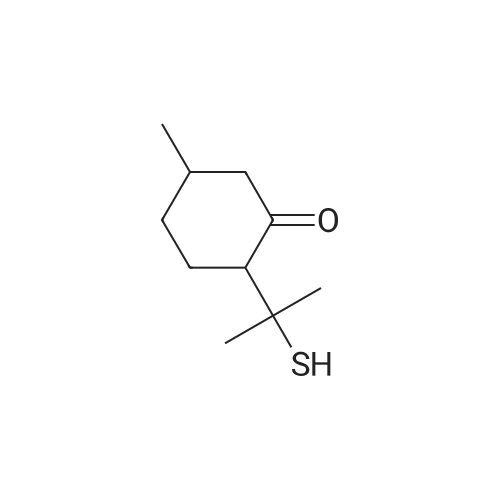
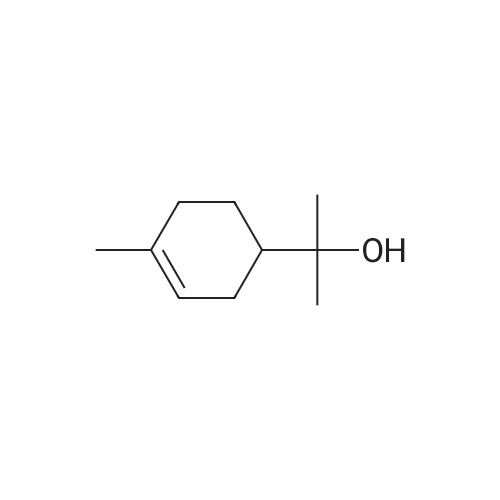
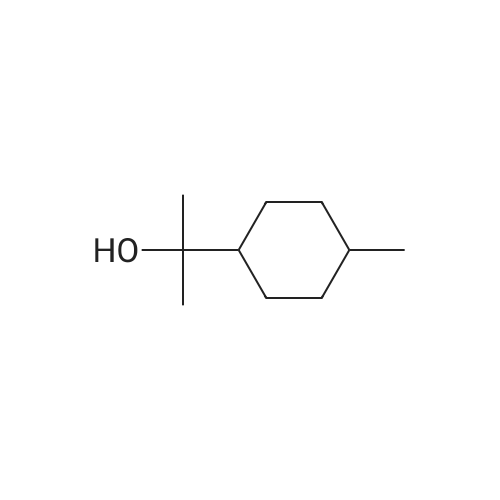
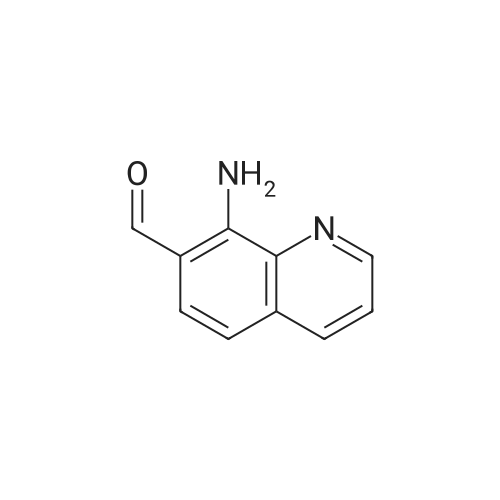
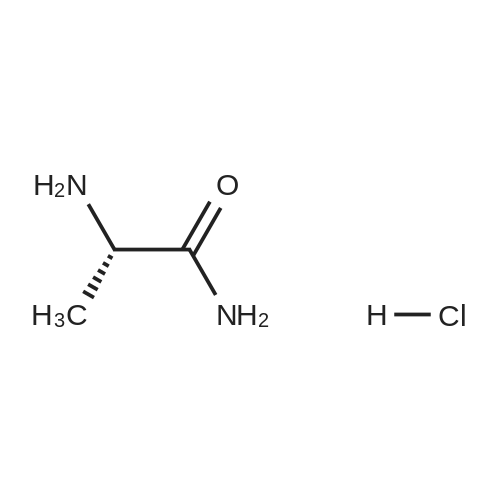
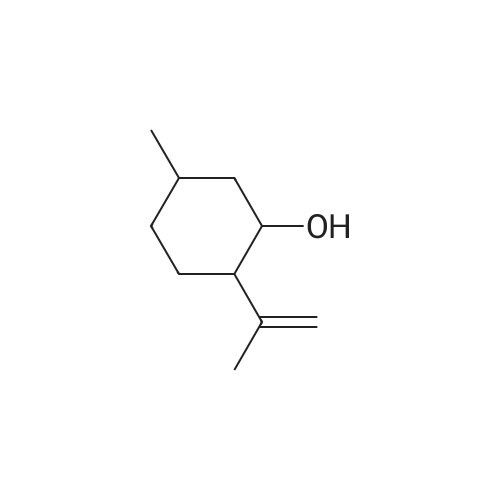
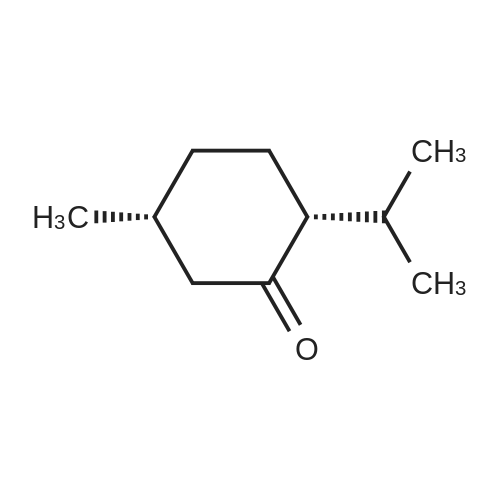
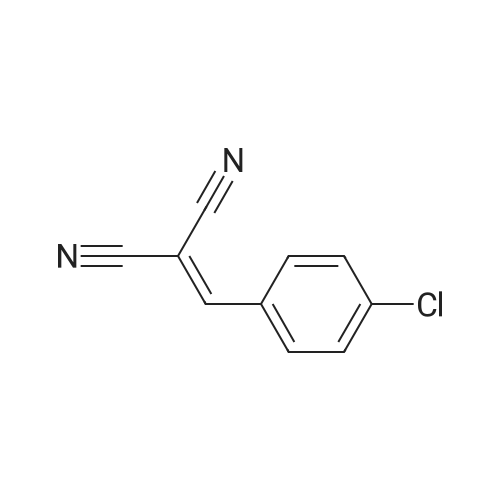
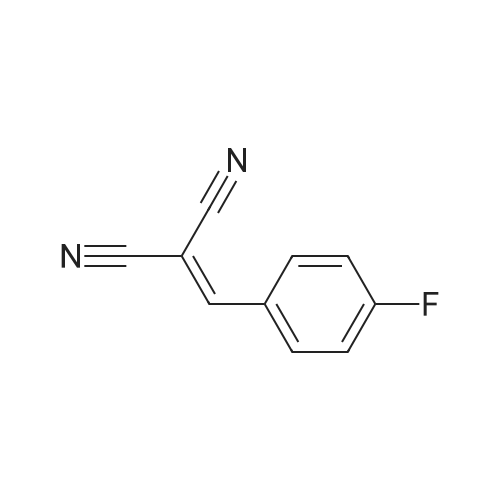
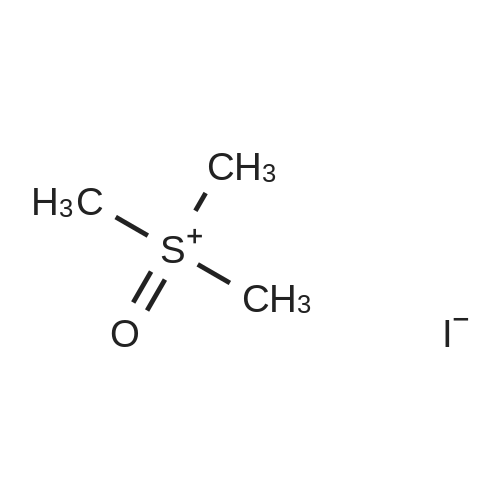
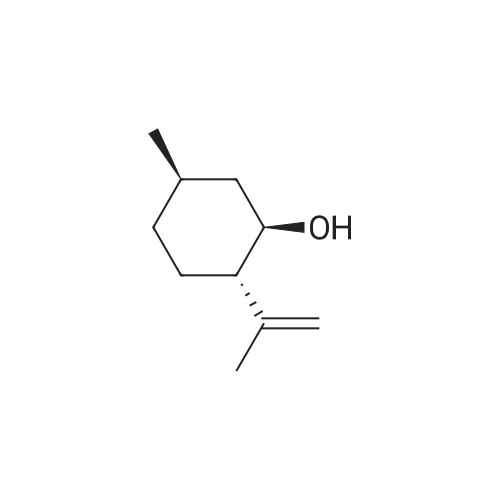
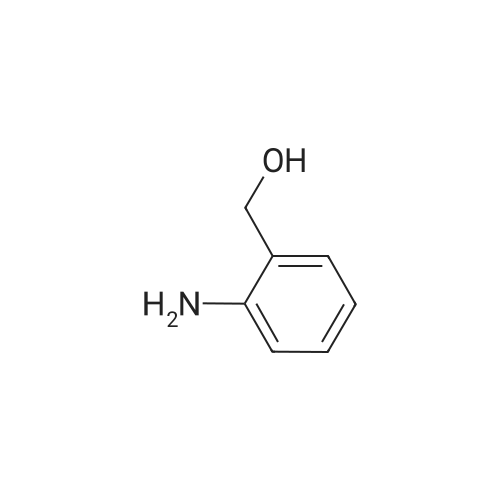
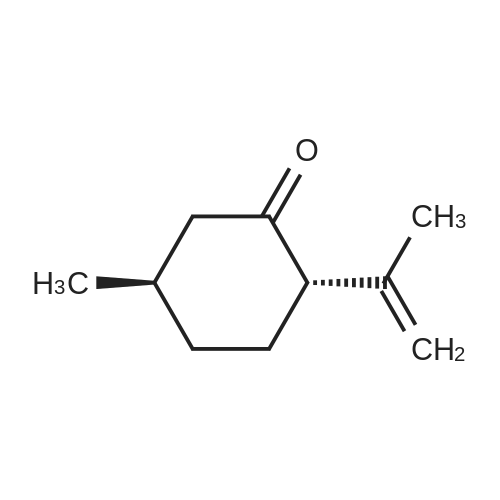
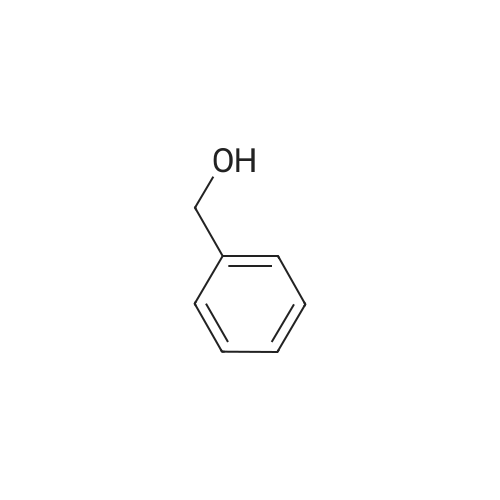
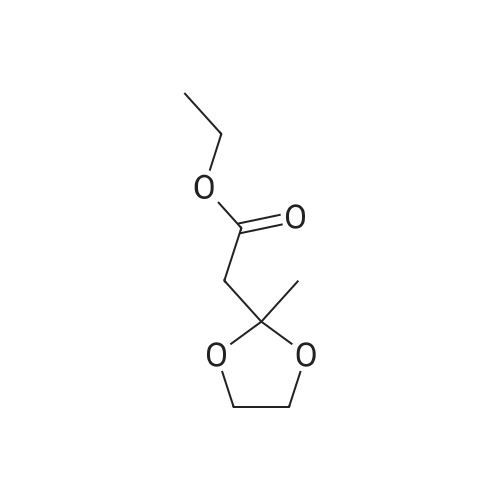
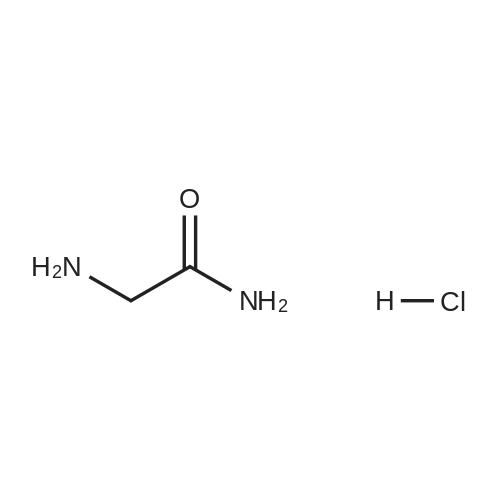
| 97% |
With dihydrogen peroxide; 1-butyl-3-methylimidazolium Tetrafluoroborate for 2h; Heating; |
|
| 97% |
With pyridine; 1‐methyl‐2‐azaadamantane‐N‐oxyl; 1-chloro-1λ3-benzo[d][1,2]iodaoxol-3(1H)-one In ethyl acetate at 20℃; for 5h; |
|
| 96% |
With trichloroisocyanuric acid; silica gel; potassium bromide In dichloromethane at 20℃; for 3h; |
|
| 95% |
With 3,5-dimethylpyrazolium fluorochromate(VI) at 20℃; for 0.0333333h; |
|
| 95% |
With 1-hydroxy-3H-benz[d][1,2]iodoxole-1,3-dione In ethyl acetate at 80℃; |
|
| 94% |
With aluminum oxide; pyridinium chlorochromate In hexane for 5h; Ambient temperature; |
|
| 94.4% |
With ruthenium(IV) oxide; tetrabutylammomium bromide; tetra(n-butyl)ammonium hydroxide In water; acetonitrile electrooxidation on Pt electrodes; |
|
| 93% |
With pyridine; oxygen In toluene at 80℃; for 2h; |
|
| 93% |
With 1-hydroxy-3H-benz[d][1,2]iodoxole-1,3-dione In ethyl acetate for 6h; Heating; |
|
| 93% |
With pyridine; 2,2,6,6-tetramethyl-piperidine-N-oxyl; 1,2-Dichloro-3-iodobenzene In chloroform at 50℃; for 0.75h; |
|
| 92% |
With peracetic acid; sodium bromide In ethyl acetate at 29.9℃; for 2h; |
|
| 92% |
With bis(quinuclidine)bromine(I) bromide In dichloromethane; water for 7h; Ambient temperature; |
|
| 92% |
With PFC In dichloromethane at 25℃; for 6h; |
|
| 92% |
With air In toluene at 110℃; for 4h; atmospheric pressure; |
|
| 91% |
With peracetic acid In tetrachloromethane; dichloromethane at 0℃; for 1h; |
|
| 91% |
With imidazolium fluorochromate In acetonitrile at 20℃; for 3h; |
|
| 90% |
In various solvent(s) at 100℃; for 28h; |
|
| 90% |
With IrH5(P-(i-Pr)3)2 In various solvent(s) at 100℃; for 28h; |
|
| 90% |
With Me-IBX In acetone at 20℃; for 1.5h; |
|
| 89% |
With dihydrogen peroxide; tetra(n-butyl)ammonium hydrogensulfate In <i>tert</i>-butyl alcohol at 90℃; for 0.5h; |
|
| 89% |
With N-Bromosuccinimide; fipronilβ-cyclodextrin In methanol; water; acetone at 20℃; for 12h; |
|
| 89% |
With pivaloyl chloride; dimethyl sulfoxide; triethylamine In dichloromethane at -78 - 20℃; for 1h; |
|
| 89% |
With oxygen; sodium carbonate In water for 2.66667h; Reflux; |
General procedure: A mixture of alcohol (1 mmol), Na2CO3(2 mmol), and Fe3O4SiO2/CuO nanocatalyst (0.04 g) in water was stirred under oxygen at reflux condition. After reaction completion, the catalyst was separated from the reaction mixture by external magnetic field, washed with hot EtOAc (2 × 5 mL), and dried for consecutive reaction runs. Then, the filtrate was cooled to room temperature, quenched with 2 MHCl aqueous solution, filtered, and extracted with dichloromethane. The solvent was evaporated and the organic layer dried over anhydrous Na2SO4. Evaporation of the solvent followed by column chromatography on silica gel (n-hexane/ethyl acetate 9:1 as v/v%) afforded the pure products. |
| 88% |
With oxygen; caesium carbonate In water for 3h; Reflux; |
General procedure
General procedure: All reactions were performed in a glass flask slurry reactor connected to an O2 tube for atmosphere control and a condenser for reflux condition. A mixture of alcohol (1mmol), Cs2CO3 (0.5mmol) and 2Au/1CuO-ZnO (0.05g) in water was stirred under oxygen atmosphere in a slurry reactor at total reflux condition. Then the catalyst was recovered by filtration, washed two times with 5ml hot EtOAc, and dried for consecutive reaction runs. The filtrate was quenched with 2M HCl aqueous solution, extracted with EtOAc three times and dried over anhydrous MgSO4. Evaporation of the solvent followed by column chromatography on silica gel afforded the pure products (Table3). |
| 87% |
With sodium hydroxide; potassium perrhuthenate for 1h; |
|
| 87% |
With aluminium trichloride; benzyltriphenylphosphonium periodate In acetonitrile for 8h; Heating; |
|
| 87% |
With N-Bromosuccinimide In methanol; water at 20℃; for 6h; Green chemistry; |
General Procedure for Oxidation of Alcohols
General procedure: Aromatic alcohol (1-2 mmol) was dissolved in methanol (or acetone in somecases, 2-4 mL) at room temperature, followed by addition of the aqueous solutionof N-bromosuccinimide (1.5 eq.=alcohol) with continuous stirring. GMP-b-CD (3,100mg=mmol of alcohol) were added in the reaction mixture and stirring was continued.Progress of the reaction was monitored by TLC until the reaction was completed(4-6 h). GMP was separated by filtration after completion of the reaction. Thereaction mixture was extracted with ethyl acetate (45 mL), combined organiclayers were dried over Na2SO4, and solvent was removed under reduced pressure.The product was further purified by flash column chromatography and analyzedby NMR spectroscopy. |
| 86.5% |
With acetone at 115℃; |
|
| 86% |
With sodium bromite In acetic acid for 5h; Ambient temperature; |
|
| 86% |
With fipronilβ-cyclodextrin; 1-hydroxy-3H-benz[d][1,2]iodoxole-1,3-dione In water; acetone at 20℃; for 12h; |
|
| 86% |
With copper phthalocyanine; tetra-n-butylammonium hydrogen monopersulfate In water at 85℃; Green chemistry; |
|
| 85% |
With potassium chlorochromate In acetone for 3h; |
|
| 85% |
With 4-methylmorpholine N-oxide In dichloromethane for 1h; Ambient temperature; |
|
| 85% |
With dihydrogen peroxide In benzene at 70℃; for 3h; |
|
| 85% |
With oxygen In water at 120℃; for 3h; |
|
| 85% |
With bis(chlorine)-1,4-diazabicyclo[2.2.2]octane at 180℃; for 0.0833333h; microwave irradiation; |
|
| 85% |
With tert.-butylhydroperoxide; CrO3(3-)*La(3+) at 90℃; for 1h; neat (no solvent); |
|
| 84% |
With palladium diacetate; potassium carbonate; triphenylphosphine In toluene for 15h; Heating; |
|
| 84% |
With silica gel; bis(trimethylsilyl)chromate for 0.05h; Irradiation; |
|
| 82% |
With pyridine; trichloroisocyanuric acid In acetone for 0.333333h; |
|
| 80% |
With zinc(II) chlorosulphate In dichloromethane for 2.5h; Ambient temperature; |
|
| 80% |
With pyridine; methyl-phenyl-thioether; N,N,N,N-tetraethylammonium tetrafluoroborate In acetonitrile electrochemical reaction: Pt-anode, Pt-cathode, undivided cells, 3F/mol of electrity, 100 mA; |
|
| 80% |
With 1-butyl-3-methylimidazolium chloride; 1-hydroxy-3H-benz[d][1,2]iodoxole-1,3-dione at 20℃; for 8h; |
|
| 80% |
With N-Bromosuccinimide; ammonium chloride In water; acetonitrile at 80℃; for 2.5h; |
|
| 80% |
With air; bis(salicylideniminato-3-propyl)methylamino-cobalt(III); 2,6-dimethoxy-p-quinone In toluene at 100℃; for 2h; |
|
| 79% |
With aluminum oxide; sodium bromite In dichloromethane for 24h; Ambient temperature; |
|
| 78% |
With 3-carboxypyridinium trifluoroacetochromate In dichloromethane at 20℃; for 1.5h; |
|
| 78% |
With 2-chloro-1,3-dimethylimidazolinium chloride; dimethyl sulfoxide; triethylamine In dichloromethane at 20℃; for 24h; |
|
| 76% |
With oxygen; isobutyraldehyde In acetonitrile for 3h; Ambient temperature; |
|
| 76% |
With <bis(salicylidene-N-methyl 3-hydroxypropionate)>cobalt; oxygen; isobutyraldehyde In acetonitrile |
|
| 75% |
With 1-[4-(diacetoxyiodo)benzyl]-3-methylimidazolium BF4; 1-ethyl-3-methylimidazolium tetrafluoroborate In acetonitrile at 30℃; for 18h; |
|
| 75% |
With 1,1,3,3-tetramethylguanidinium fluorochromate In dichloromethane for 0.00277778h; Microwave irradiation; |
2.4. General Procedure for the Oxidation under Microwave Irradiation
General procedure: The substrate (1mmol) and 1.5-2 mmol oxidant were mixed. To this mixture 0.5 mL CH2Cl2 was added. The mixture was subjected to microwave irradiation (1000 W). Upon completion of the reaction, extraction with ether (3 × 25mL) and evaporation of the solvent gave the corresponding carbonyl compounds. The products formed were analyzed by their 2,4-dinitrophenylhydrazone derivatives.The precipitated 2,4-DNP was filtered off, weighed, and recrystallized from ethanol. |
| 74% |
With HMTAB; silica gel In water at 20℃; for 0.0833333h; |
|
| 74% |
With aluminum oxide; quinolinium monofluorochromate(VI) In hexane for 2.5h; Ambient temperature; |
|
| 72% |
With oxygen; 2-ethoxycarbonyl-1-cyclopentanone In acetonitrile at 60 - 70℃; |
|
| 71% |
With oxygen; sodium acetate In water; dimethyl sulfoxide at 80℃; for 4h; |
|
| 70% |
With tetrabutyl-ammonium chloride; dihydrogen peroxide; potassium carbonate In tetrahydrofuran for 120h; Ambient temperature; |
|
| 70% |
With quinolinium monofluorochromate(VI) In dichloromethane for 4h; Heating; |
|
| 70% |
With hexamethylenetetrammonium fluorochromate In N,N-dimethyl-formamide at 20℃; for 4h; |
|
| 70% |
With iodine; potassium carbonate; potassium iodide at 90℃; for 0.75h; |
|
| 69% |
With 2,6-dimethylpyridine; methyl octyl sulfide; tetraethylammonium bromide In benzonitrile |
|
| 69% |
With 1-hydroxytetraphenylcyclopentadienyl(tetraphenyl-2,4-cyclopentadien-1-one)-μ-hydrotetracarbonyldiruthenium(II) In 1,3,5-trimethyl-benzene at 165℃; for 36h; Inert atmosphere; |
|
| 68% |
With [MnIII(2-((2-(2-(2-(2-hydroxybenzylideneamino)phenylamino)propylamino)phenylimino)methyl)phenolato)]Cl; dihydrogen peroxide In acetonitrile for 4h; Reflux; |
|
| 66% |
With potassium chlorochromate on alumina In dichloromethane for 73h; Ambient temperature; |
|
| 65% |
With 3,5-dimethylpyrazolium fluorochromate(VI) In dichloromethane at 20℃; for 2h; |
|
| 60% |
With oxygen; benzaldehyde In 1,2-dichloro-ethane at 80℃; for 36h; |
|
| 58% |
With pyridine; N-hydroxyphthalimide; sodium perchlorate In acetonitrile electrolytical oxidation, anode potential 0.85 V; |
|
| 54% |
With (carbonyl)(chloro)(hydrido)tris(triphenylphosphine)ruthenium(II); oxygen In toluene at 90℃; for 18h; Molecular sieve; Sealed tube; |
|
| 52% |
With tert.-butylhydroperoxide In benzene at 60℃; for 12h; |
|
| 50% |
With oxygen; nitrosonium tetrafluoroborate In dichloromethane at 20℃; for 16h; |
|
| 48% |
With tetra-N-butylammonium tribromide In acetonitrile at 20℃; for 72h; Irradiation; |
2.2 General experimental procedure for the photocatalytic oxidation of alcohol
General procedure: In a 50ml Pyrex round-bottom flask, a mixture of alcohol (1mmol), TBATB (10-20mg, 0.02-0.04mmol) in 10ml of CH3CN was exposed to blue or violet light LED irradiation at room temperature under an air atmosphere with stirring. The progress of the photocatalytic oxidation reaction was monitored by TLC on silica gel plates. The reaction mixture externally irradiated until the alcohol was completely consumed. |
| 45% |
With [(C18H37)2(CH3)2N]3[SiO4H(WO5)3]; dihydrogen peroxide In water; ethyl acetate at 79.84℃; for 10h; |
|
| 33.9% |
With tert.-butylhydroperoxide at 70℃; for 5.5h; |
|
| 28% |
With tert.-butylhydroperoxide In hexane; water at 50℃; for 16.5h; |
|
| 26% |
With tert.-butylhydroperoxide; C14H30Cl2FeN4(1+)*F6P(1-) In [D3]acetonitrile; water; water-d2 at 27℃; for 18h; Inert atmosphere; Schlenk technique; Green chemistry; |
|
| 20% |
With dihydrogen peroxide In toluene at 75℃; for 7h; |
|
| 19.2% |
With acetone In benzene at 80℃; for 8h; other temperature; |
|
| 19% |
With tetrabutylammomium bromide; 1-hydroxy-3H-benz[d][1,2]iodoxole-1,3-dione In dichloromethane; water at 20℃; for 4h; |
|
| 19% |
With dipyridinium dichromate; adogen 464; dihydrogen peroxide; sodium carbonate In various solvent(s) for 24h; Heating; |
|
|
With chromium(VI) oxide; acetic acid at 0℃; inactive p-menthanone-(3) from thymomenthol; |
|
|
With chromic acid l-menthone; |
|
| 82 % Chromat. |
With N-iodo-succinimide; tetra-(n-butyl)ammonium iodide In dichloromethane for 3h; Ambient temperature; |
|
|
With crosslinked poly-4-vinylpyridine hydrobromide In acetonitrile at 50℃; for 21.5h; electric current, 60mA, 10-30V; Yield given; |
|
|
With phosphorus pentoxide; dimethyl sulfoxide; triethylamine 1.) CH2Cl2, from 0 deg C to RT, 30 min, 2.) CH2Cl2, 0 deg C, 30 min; Yield given. Multistep reaction; |
|
| 97 % Chromat. |
With <MoO(O2)2C5H4N(O)COO>Bu4N In 1,2-dichloro-ethane at 50℃; for 9h; |
|
|
With chromic acid |
|
|
With ruthenium trichloride; pinane hydroperoxide; <i>tert</i>-butyl alcohol In chlorobenzene for 24h; Ambient temperature; Yield given; |
|
| 92 % Chromat. |
With tert.-butylhydroperoxide In dichloromethane; 1,2-dichloro-ethane at 40℃; for 12h; |
|
|
With aluminum oxide; bromine In dichloromethane for 1h; |
|
|
With SiW11Zn; dihydrogen peroxide In water at 89.85℃; for 9h; |
|
| 80 % Chromat. |
With CrO3/silica gel In various solvent(s) at 40℃; for 4h; |
|
|
With quinoxalinium fluorochromate In dichloromethane for 0.00416667h; Microwave irradiation; |
|
| 89 % Chromat. |
With chromium(VI) oxide for 0.05h; microwave irradiation; |
|
|
With polystyrene-supported hypervalent iodine(V) reagnet In 1,2-dichloro-ethane at 85℃; for 1h; |
|
| 90 % Chromat. |
With Oxone; sodium chloride In water; ethyl acetate at 20℃; for 1.5h; |
|
| 82 % Chromat. |
With Oxone; 2-Iodobenzoic acid; tetra(n-butyl)ammonium hydrogensulfate In ethyl acetate at 70℃; for 6h; |
|
|
With 1,3,5-trichloro-2,4,6-triazine; dimethyl sulfoxide; triethylamine In tetrahydrofuran at -30 - 20℃; for 1.5h; |
|






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






 HazMat Fee +
HazMat Fee +

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping